角质形成细胞中异常的RNA剪接往往会导致皮肤炎症障碍等疾病。特应性皮炎( Atopic dermatitis,AD)(又称遗传过敏性皮炎)和银屑病都是慢性复发性炎症性皮肤病。尽管角质形成细胞对T细胞源性细胞因子的异常反应是特应性皮炎和银屑病病理的固有特征,但它们是否具有调节角质形成细胞炎症的共同机制尚不清楚。

2022年10月21日,华东师范大学赖玉平团队及清华大学/上海交通大学董晨团队合作在Nature Immunology(IF=31)发表了题为“IL-17D-induced inhibition of DDX5 expression in keratinocytes amplifies IL-36R-mediated skin inflammation”的研究论文,该研究发现特应性皮炎和银屑病患者来源的角质形成细胞中RNA解旋酶DDX5下调,且DDX5通过调节IL-36R pre-mRNA的可变剪接来调控IL-36R的表达进而促进角质形成细胞炎症应答。
为了证明DDX5在皮肤炎症中的作用,作者首先分析了GEO数据库中健康人群、特应性皮炎和银屑病患者皮肤的RNA-Seq和单细胞测序(scRNA-Seq)结果,发现DDX5 mRNA的表达在特应性皮炎和银屑病患者皮肤损伤中显著降低(图1)。
免疫荧光染色进一步证实DDX5蛋白主要在健康对照者皮肤角质形成细胞中高表达,而在特应性皮炎和银屑病患者皮肤损伤角质形成细胞中的表达显著降低。另外,在OVA或MC903(卡泊三醇)诱导的特应性皮炎小鼠和咪喹莫特(IMQ)诱导的银屑病小鼠皮肤损伤角质形成细胞中DDX5 mRNA和蛋白的表达也被抑制(图2)。为了证明DDX5在特应性皮炎和银屑病中的作用,他们构建了在角质形成细胞中特异敲除Ddx5 (Ddx5∆KC) 的条件小鼠。与Ddx5fl/fl同窝小鼠相比,Ddx5∆KC小鼠对MC903和IMQ更敏感,主要表现在:MC903诱导的Ddx5∆KC特应性皮炎小鼠皮肤上有明显增厚的鳞片和更多的斑块,皮肤损伤中与特应性皮炎致病相关的关键因子Il4、Il13、Ccl11、Ccl17 和 Ccl22的表达显著增加,皮肤损伤中浸润的CD45+免疫细胞增多;IMQ诱导的Ddx5∆KC银屑病小鼠皮肤上斑块增多,表皮层棘层增厚,皮肤损伤中与银屑病致病相关的关键细胞因子、趋化因子以及免疫细胞的浸润也明显增多,种种实验结果共同表明:角质形成细胞中DDX5的缺失会诱发皮肤炎症。
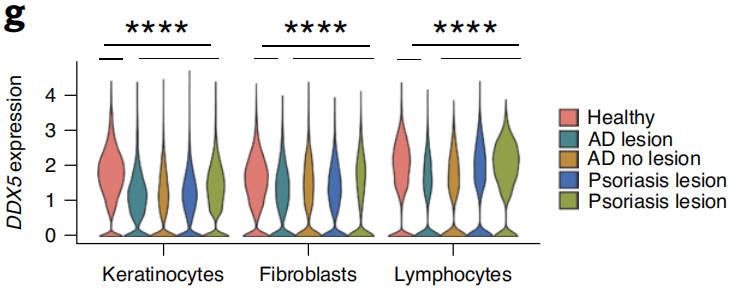
图1 健康成人(n=5)及AD (n = 4)和银屑病(n = 3)患者皮肤scRNA-seq中角质形成细胞、成纤维细胞和淋巴细胞中DDX5 mRNA的表达量示意图。
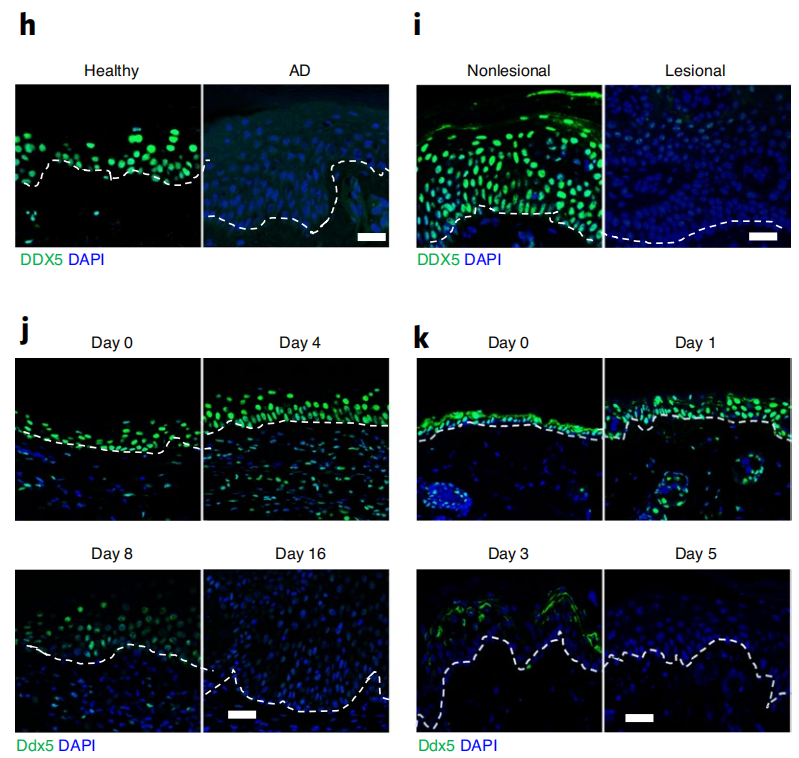
图2 免疫荧光分析3例AD患者的健康皮肤(n = 3)和皮肤损伤皮肤(h)、3例银屑病患者的非皮肤损伤和皮肤损伤皮肤(i)或MC903处理的野生型小鼠(j)或IMQ处理的野生型小鼠(k)的皮肤损伤皮肤中的DDX5+细胞。
随后研究显示,与未处理的小鼠相比,MC903或IMQ处理的野生型小鼠皮肤中IL-17D的表达量稳步增加。进一步的实验结果表明IL-17D是抑制角质形成细胞中DDX5表达的关键细胞因子,它通过激活CD93-p38 MAPK-AKT-SMAD2/3信号通路抑制DDX5的表达。
另外,研究人员通过体外细胞实验证明DDX5的敲减能选择性增强角质形成细胞对IL-36的应答。作者对WT vs DDX5–/–角质形成细胞、Ddx5fl/fl vs Ddx5∆KCAD小鼠皮损和Ddx5fl/fl vs Ddx5∆KC银屑病小鼠皮损进行RNA-Seq,综合分析三个RNA-Seq数据后发现DDX5主要调节与白细胞迁移、正向调节细胞因子产生和细胞趋化作用相关的基因的表达,这些均是导致特应性皮炎和银屑病的关键因素(图3)。另外DDX5的下调还能上调IL-36R在角质形成细胞、Ddx5∆KC特应性皮炎和银屑病皮肤损伤中的表达。并且,在DDX5–/–角质形成细胞中敲低IL-36R能明显抑制DDX5–/–角质形成细胞对IL-36的应答,证明DDX5的缺失通过上调IL-36R的表达促进角质形成细胞对IL-36的应答。
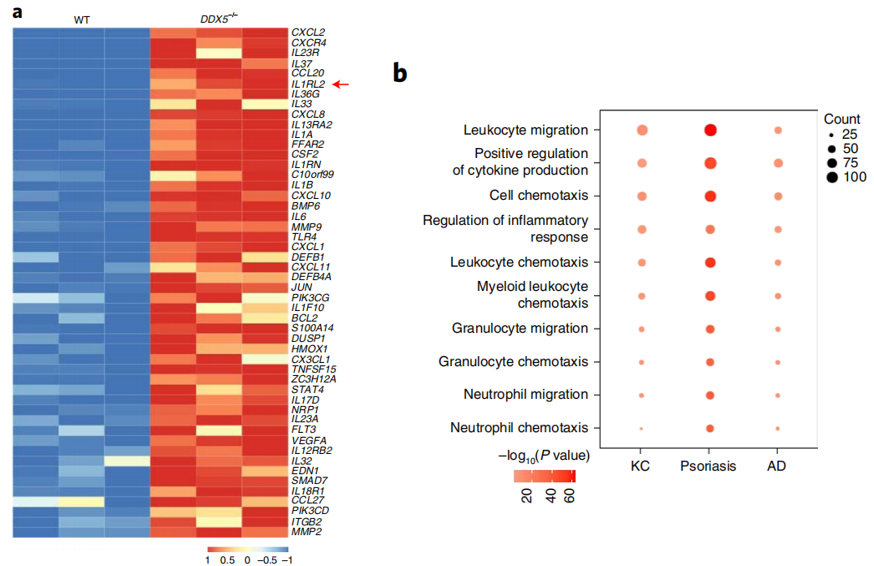
图3 a,通过RNA-seq分析,显示野生型和DDX5-/- HaCaT细胞(n = 3)中被100 ng/ml IL-36γ刺激4小时的前50个上调基因的热图。红色箭头指向IL1RL2 (IL-36R)。b,从MC903处理的Ddx5∆KC (n = 4)和Ddx5fl/fl小鼠(n = 3)或IMQ处理的Ddx5∆KC (n = 3)和Ddx5fl/fl小鼠(n = 3)中获得的导致图a和整个损伤皮肤的RNA-seq GO富集分析的前十个新陈代谢类别。
该研究中使用的 IL-36γ (CR58)来自近岸蛋白,近岸蛋白还提供多种靶点蛋白和细胞因子,以过硬的品质和齐全的种类被广大科研工作者所青睐,欢迎大家咨询选购!
接下来,该研究发现DDX5可能通过调节IL-36R pre-mRNA的可变剪接来调控IL-36R的表达进而促进角质形成细胞炎症应答的机制。利用DNA-PAGE检测内源性IL-36R的剪接和IL-36R miniGene报告系统也证实DDX5确实能调节人源IL-36R pre-mRNA的3号外显子跳跃或鼠源IL-36R pre-mRNA的6号外显子跳跃,均使IL-36R pre-mRNA的翻译提前终止,产生含IL-36R胞外段的可溶性蛋白(sIL-36R)(图4)。后续实验证明DDX5与SF2协同调节IL-36R剪接,在角质形成细胞中产生sIL-36R。
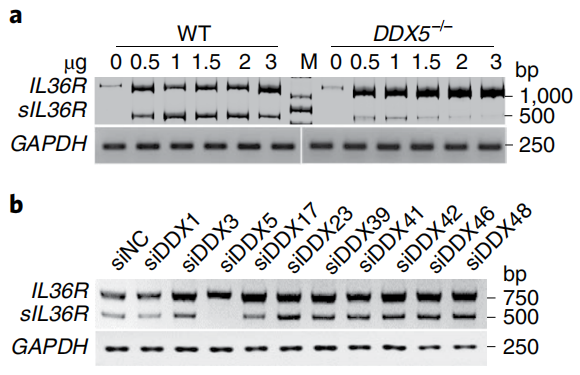
图4 a转染0-3 µg人源IL36R报告基因序列片段的野生型和DDX5-/- HeLa细胞中IL36R和sIL36R含量的电泳图。b,靶向DDX1、DDX3、DDX5、DDX17、DDX23、DDX39、DDX41、DDX42、DDX46和DDX48的siRNA转染后NHEK细胞中IL36R和sIL36R含量的电泳图。
接下来,该研究通过免疫共沉淀和流式细胞术证明sIL-36R与IL-36R竞争结合它们的配体IL-36来抑制IL-36-IL-36R信号通路(图5和图6)。在角质形成细胞中表达sIL-36R能抑制角质形成细胞对IL-36的免疫应答.该研究通过给Ddx5∆KC特应性皮炎或银屑病小鼠注射sIL-36R或在角质形成细胞中回补sIL-36R验证sIL-36R能抑制皮肤炎症,减轻疾病症状,证明特应性皮炎和银屑病皮损中sIL-36R的减少是Ddx5∆KC小鼠皮肤炎症加重的诱因。
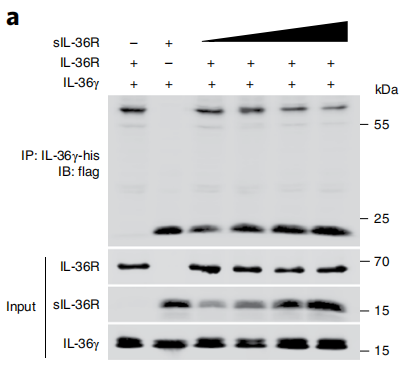
图5 用含有Flag标签的sIL-36R或IL-36R的质粒转染的HeLa细胞的总细胞裂解物中Il -36γ结合IL-36R和sIL-36R的免疫沉淀结果电泳图。
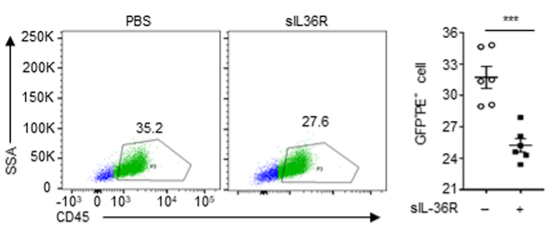
图6 流式细胞术分析HeLa细胞中IL-36γ和IL-36R在IL-36R过表达和不过表达情况下的相互作用结果数据分析图(n = 6)。
综上所述,该研究揭示IL-17D- CD93抑制角质形成细胞中DDX5的表达可以加重皮肤炎症,提示IL-17D和DDX5可作为炎症性皮肤病的治疗的潜在靶点。另外,关于sIL-36R的研究为RNA剪接异常如何参与皮肤炎症的发病机制提供了新的机制阐述,也为特应性皮炎和银屑病的治疗提供了潜在的治疗方法。
该研究中使用的2×Taq Master Mix (E005)来自近岸蛋白,近岸蛋白的分子产品已经被广泛的应用于基础科研的各个领域,许多实验室在近岸蛋白产品的支持下陆续发表了很多高分文章,近岸蛋白是您科研道路上的好帮手!
近岸蛋白相关产品
|
货号 |
产品名称 |
|
2×Taq Master Mix |
|
|
Taq DNA Polymerase (with dNTP) |
|
|
5×Multiplex PCR Mix |
|
|
NovoRec® plus One step PCR Cloning Kit |
|
|
NovoScript®Plus All-in-one 1st Strand cDNA Synthesis SuperMix (gDNA Purge) |
|
|
NovoStart®SYBR qPCR SuperMix plus |
|
|
Recombinant Mouse IL-36 gamma |
|
|
Recombinant Human/Mouse/Rat Activin A |
|
|
Recombinant Human EGF |
|
|
NovoNGS® DNA Library FlashPrep Kit for Illumina® |
|
|
NovoNGS® mRNA Magnetic Isolation Kit |
论文链接:
https://www.nature.com/articles/s41422-022-00735-6